
随着经济的高速发展,我国对医疗健康的需求越来越大,医疗器械的发展前景也较为广阔。与此同时,国家对医疗器械的监管也在不断完善。1989年我国开始引入医疗器械市场准入的概念,1992年借鉴欧洲监管模式,我国启动医疗器械产品安全认证工作……
我国的医疗器械监管模式借鉴了美国和欧盟的监管经验,与大多数国家一样对医疗器械实行分类管理,同时既有上市前审批,又有上市后监管。本文将对医疗器械法规进行简单介绍。

医疗器械。
医疗器械的监管形式有产品监督和企业监督两种,产品监督包括上市前许可(产品注册审批)和上市后监督(不良事件监测、市场监督);企业监管则是指生产企业、经营企业的许可、备案和日常监督。
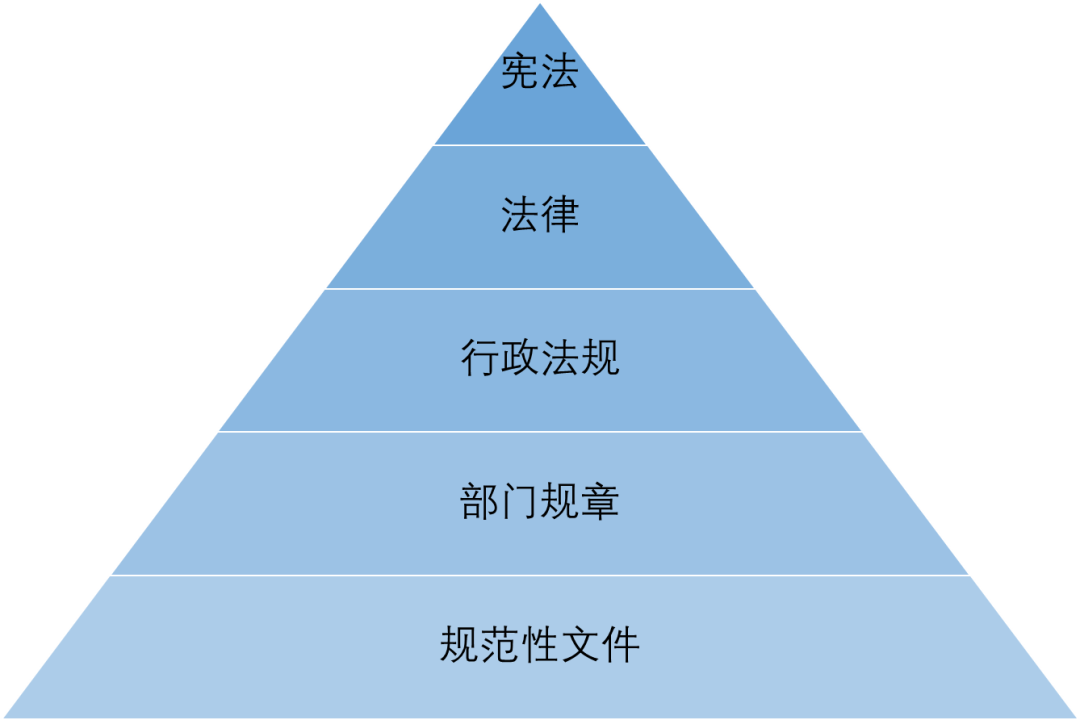
国家药品监督管理局是全国医疗器械的主管机构,负责制定医疗器械监督管理、质量管理规范的政策、规划并监督实施;负责医疗器械行政监督和技术监督;负责医疗器械注册和监督管理,组织开展医疗器械不良事件监测,组织实施分类管理制度。国家药品监督管理局内设政策法规司、医疗器械监管司、稽查局等机构,负责相关医疗器械管理工作。此外,我国采用多级监督模式,下设省级、市级和县级药品监督管理机构。 中国医疗器械监管法律体系可以参照下图的金字塔式结构。就第二层的法律而言,中国虽制定有《食品安全法》和《药品管理法》,但尚未制定专门针对医疗器械监管的法律。 行政法规层面是由国务院发布的《医疗器械监督管理条例》(以下简称《条例》),通常包括上市前和上市后监管要求。部门规章层面是由国家医疗器械监管部门发布的部门法规,指导医疗器械制造商在设计、生产、注册登记、销售、售后等各个环节符合法规要求,如《医疗器械注册管理办法》。规范性文件由国务院下属部委制定,如《关于印发医疗器械生产质量管理规范(试行)的通知》。 中国医疗器械监管法律体系的金字塔式结构。 国务院2000年制定了《医疗器械监督管理条例》,2014年、2017年分别做了全面修订和部分更改,其对保障医疗器械质量安全、推动行业健康发展有着重要作用。 之前的文章中所介绍的医疗器械分类也是按照《条例》进行的,是医疗器械注册、生产、经营、使用全过程监管的重要基础。 医疗器械的分类。 1.医疗器械产品注册与备案制度 根据《条例》,第一类医疗器械需要备案人在设区的市级人民政府药品监督管理部门备案;申请第二类医疗器械产品注册需要注册申请人在省、自治区、直辖市人民政府药品监督管理部门注册;申请第三类医疗器械产品注册需要注册申请人在国务院药品监督管理部门注册。 2.医疗器械生产许可制度 第一类医疗器械的生产要在设区的市级人民政府药品监督管理部门予以备案;第二类、第三类医疗器械的生产需经省、自治区、直辖市人民政府药品监督管理部门审批后发放医疗器械生产许可证。医疗器械生产许可证有效期为5年。有效期届满需要延续的,依照有关行政许可的法律规定办理延续手续。 3.医疗器械经营许可制度 经营第一类医疗器械不需要许可和备案;经营第二类医疗器械的企业需要向设区的市级人民政府负责药品监督管理的部门备案,按照国务院药品监督管理部门的规定对产品安全性、有效性不受流通过程影响的第二类医疗器械可以免于备案;经营第三类医疗器械的企业需要经设区的市级人民政府药品监督管理部门审批并给予医疗器械经营许可证。医疗器械经营许可证有效期为5年。有效期届满需要延续的,依照有关行政许可的法律规定办理延续手续。 医疗器械监管体系对推动行业健康发展有着重要作用。 除了《条例》,在临床、注册、生产、经营、使用的各个环节中还有其他法律文件,它们一起对医疗器械的全生命周期和全过程进行监管。采用科学的医疗器械管理体系才能帮助促进我国医疗器械产业持续不断地创新发展,同时也保证人民的生命健康安全。现行法律体系

上海市松江区莘砖公路668号双子楼A栋1003室
电话:18964878976
展会咨询QQ:515616785
传真:021-31078232



